从“筛选”到“设计”:人工智能助力核酸适配体的发现
发布时间:2026-03-23
引言:从"大自然"到"实验室"的精准识别
在生命科学的微观世界里,"精准识别"是诊断与治疗的共同基石。长期以来,抗体凭借出色的特异性与亲和力,被视为生物医学领域的"黄金标准"。然而,随着精准医疗的发展,一类新型识别分子--核酸适配体(nucleic acid aptamer)正在快速走入大众视野,并在多个场景中展现出独特优势。
核酸适配体通常由25~80个碱基组成的单链DNA或RNA构成。它们就像一条条富有韧性的"分子丝带",可以通过"折纸"般折叠形成复杂空间构象,从而实现"钥匙入锁"般的精准识别:不仅能够结合蛋白质、小分子,还可识别病毒颗粒甚至完整细胞。与传统抗体相比,核酸适配体具有分子质量小、热稳定性强、易于化学合成与修饰、免疫原性低等特点,因此在肿瘤诊疗、生物传感、食品安全与环境监测等方向被寄予厚望[1-2]。
然而,它面临的最大挑战是:核酸适配体虽然好用,应用前景广阔,却并不好找。
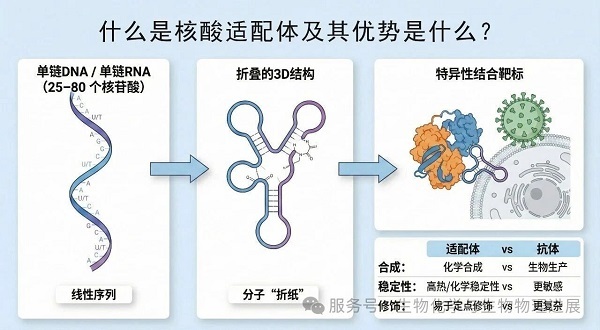
图1 核酸适配体:从线性序列到三维折叠,实现高特异性识别
现状:从"大海捞针"到"数智化"飞跃
自1990 年指数富集配体系统进化技术(SELEX)提出以来,核酸适配体的发现长期依赖实验室的"物理筛选"[3]。其核心流程可以概括为:①准备一个包含海量随机序列的核酸文库;②让文库与目标靶标结合;③保留"结合得更好"的序列并扩增;④多轮迭代,直到富集出高亲和候选。
SELEX奠定了核酸适配体研究的技术基础,但也常被诟病为轮次多、周期长、成本高、对实验条件敏感。尤其面对更复杂的靶标(如膜蛋白、细胞/组织水平靶标、构象变化显著的体系),最终"筛出来"的结果并不总能代表真正的最优解。
近年来,高通量测序驱动的HT-SELEX显著提升了筛选效率,但也带来了一个新的难题:数据爆炸[4]。面对动辄千万量级的序列与富集轨迹,仅靠传统统计与人工经验已难以充分挖掘其中的规律。也正是在这一背景下,人工智能(AI)的加入改变了游戏规则--它让核酸适配体研发从"凭经验做实验",逐步走向"数据驱动的智能决策"。
概括来说,AI在核酸适配体领域正在做三件关键的事:预测、解释、再设计。
预测:从序列直接判断"更可能结合"的候选
深度学习可以把核酸序列当作一种"语言",从序列片段模式与富集变化中学习统计规律,并据此对候选进行排序。代表性工作如AptaNet [5],尝试融合适配体与蛋白质特征来预测适配体-蛋白质相互作用对。这类方法的实际价值在于:先用计算做"初筛",再把实验资源集中投给更可能命中的候选。
解释:不只给分数,还要回答"为什么"
核酸适配体不是简单的"字母串",其功能很大程度上取决于折叠后的空间结构。仅从序列出发的模型容易出现"看起来像、但折不出来"的候选。为此,越来越多研究引入二级结构或结构特征,并结合经典工具(如ViennaRNA/RNAfold)进行结构约束与表征,从而帮助回答[6-7]:哪些片段更可能形成稳定的茎环结构?哪些结构位点可能是关键结合核心?如何从"富集现象"进一步提炼为可解释的设计线索?
再设计:从"筛出来"走向"生成出来"
更进一步,模型不仅用于排序,还可以基于学习到的规律提出新的序列方案。近年来也出现利用早期轮次数据并结合序列与结构特征挖掘高亲和候选的深度模型(如AptaDiff 等思路)。这些进展共同指向一个更具想象力的方向:更少轮次、更短周期、更接近闭环自动化的核酸适配体发现流程[8-9]。
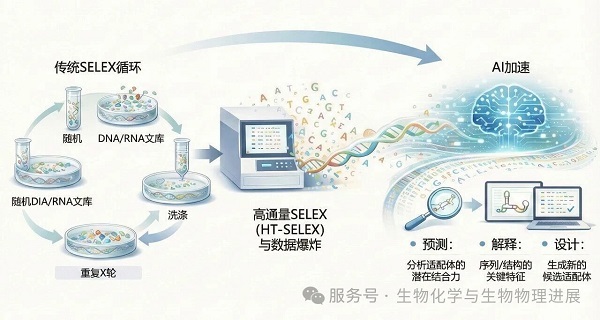
图2 SELEX→HT-SELEX→AI:核酸适配体发现从筛选走向数据驱动加速
挑战:生命"丝带"的灵动与复杂
尽管AI在生物学领域已经取得了如AlphaFold般的突破性进展,但当它走进核酸适配体这条"生命丝带"的世界,仍会遇到更深水区的难题。归纳来看,主要挑战集中在三个方面:
(1)数据的"孤岛化"
现有核酸适配体数据库(如AptaDB、UTexas Aptamer Database)为研究提供了重要资源,但整体仍存在格式不统一、元数据注释不完整、实验条件记录粒度不一致等问题。不同平台之间难以顺畅对接,导致跨数据库整合、统一建模与公平评测都十分困难[10]。
(2)极高的构象柔性
与结构相对稳定的蛋白质相比,单链核酸(尤其是RNA)具有更强的柔性与构象多样性。它的三维折叠会受到离子强度、pH、温度等环境因素显著影响,甚至同一条序列在不同条件下也可能呈现截然不同的构象与结合行为。这使得"从序列直推功能"的难度大幅上升[11]。
(3)预测与验证的断层
计算模型推荐的"高亲和力"候选序列,在实验室中往往表现并不稳定:有的候选难以折叠成预期结构,有的在真实体系里受非特异吸附、基质干扰或条件偏差影响而失效。当前仍缺少高效的"设计-验证-反馈"闭环体系,限制了核酸适配体从计算发现走向稳定转化与规模化应用的速度。
核心意义:构建一体化的计算设计蓝
针对上述瓶颈,电子科技大学的林昊团队系统梳理并评述了人工智能技术在核酸适配体研发中的应用进展。这篇文章的价值不只是"归纳已有工作",更重要的是把领域从"零散工具箱"梳理成一张可执行的"路线图",帮助读者理解:该从哪里入门、按什么路径推进、关键问题卡在哪、下一步怎么做更有效。(详情请点击阅读原文)
文章按照"数据库→虚拟筛选→结构预测→相互作用预测→生成式设计→典型应用"的逻辑,系统串联核酸适配体AI的关键环节与常用策略,尽量把"单点方法"放回到"完整工作流"中讨论,从而为初学者与跨学科研究者提供更可落地的参考路径。
重点评述了生成式AI(如GAN、VAE、Diffusion Model)在核酸适配体设计中的应用进展:模型不再只是"筛选器",而开始向"候选提出者/设计者"演进,能够在特定约束下生成更符合目标需求的新序列,为"更少轮次、更快收敛"的研发模式提供可能。
进一步讨论了如何把计算侧指标(如对接得分、结构置信度指标等)与实验侧读数(如亲和力常数、传感信号、检测限等)放在同一张"评价坐标系"里对齐,并总结影响落地的关键要点(实验条件记录、负样本构建、泛化评测、可解释性与闭环验证等),为核酸适配体从"能算"走向"能用"提供更工程化的视角。
展望:开启"端到端"智能化医疗
展望未来,核酸适配体的研发将不再只是一次性的筛选实验,而会逐步演进为可持续迭代的端到端自动化闭环系统。如图所示,这一闭环可以概括为五个连续环节:数据-训练-候选者-实验-验证。
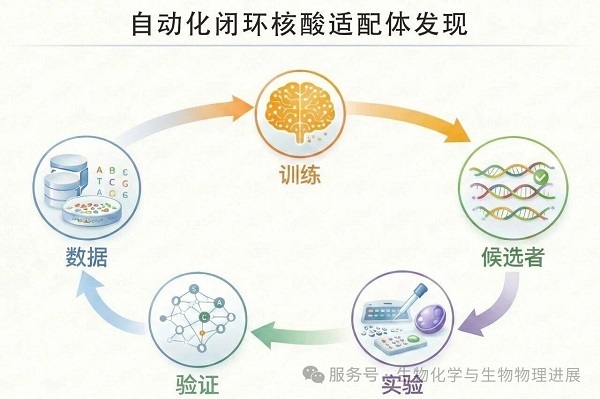
图3 自动化闭环:数据-训练-候选者-实验-验证驱动的核酸适配体研发新范式
首先,数据不再局限于最终筛选结果,而是系统汇聚HT-SELEX过程数据、结构/对接信息以及不同实验条件下的亲和力、特异性与稳定性读数,形成可复用的数据底座。随后进入训练阶段,AI模型在这些多源数据上学习"序列-结构-功能"的规律,得到更具泛化能力的预测与生成策略。基于训练后的模型,系统输出更精炼的候选者集合(例如高亲和、强特异、易修饰或更稳定的序列),把实验资源集中投向最有潜力的"少数精兵"。接着通过自动化平台(如微流控合成与筛选、高通量亲和力测定、传感读出等)完成实验,快速获得真实体系下的多维评价指标。最后在验证环节,对候选在不同条件与复杂基质中进行更严格的可重复性与泛化检验,并将验证结果回流,更新数据集与模型参数,推动下一轮更精准的训练与候选提出。
这种"实验结果反哺模型、模型再指导实验"的闭环迭代,有望在更少轮次、更短周期内获得稳定可用的核酸适配体,并能更快响应新靶标需求。随着可解释AI、主动学习以及数据标准化体系逐步成熟,核酸适配体这把生命识别的"万用钥匙"将变得更加智能与可控,也将从实验室的"精巧分子"走向临床精准诊断、即时检测与靶向治疗中的关键组件。
参考文献
[1] Chen Z, Hu L, Zhang B T, et al. Artificial intelligence in aptamer-target binding prediction. Int J Mol Sci, 2021, 22(7): 3605
[2] Fallah A, Havaei S A, Sedighian H, et al. Prediction of aptamer affinity using an artificial intelligence approach. J Mater Chem B, 2024, 12(36): 8825-8842
[3] Chatterjee O, Kaur G A, Shukla N, et al. Multifaceted arsenal in SELEX nanomedicine. Adv Colloid Interface Sci, 2025, 342: 103540
[4] Yang G, Liu W, Zhao Y, et al. Induction of binding sites for RecA aptamers by differentiated-competition capillary Electrophoresis-SELEX. Talanta, 2024, 267: 125213
[5] Emami N, Ferdousi R. AptaNet as a deep learning approach for aptamer-protein interaction prediction. Sci Rep, 2021, 11(1): 6074
[6] Sato K, Akiyama M, Sakakibara Y. RNA secondary structure prediction using deep learning with thermodynamic integration. Nat Commun, 2021, 12(1): 941
[7] Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024, 630(8016): 493-500
[8] Wang Z, Liu Z, Zhang W, et al. AptaDiff: de novo design and optimization of aptamers based on diffusion models. Brief Bioinform, 2024, 25(6): bbae517
[9] Iwano N, Adachi T, Aoki K, et al. Generative aptamer discovery using RaptGen. Nat Comput Sci, 2022, 2(6): 378-386
[10] Rigden D J, Fernández X M. The 2025 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res, 2025, 53(D1): D1-D9
[11] Ferreira L G, dos Santos R N, Oliva G, et al. Molecular docking and structure-based drug design strategies. Molecules, 2015, 20(7): 13384-13421
作者简介
刘上华:电子科技大学生命科学与技术学院博士生;研究方向:基于人工智能的核酸适配体筛选与设计。
刘入铭:电子科技大学生命科学与技术学院博士生;研究方向:核酸适配体筛选分析及生物传感器构建。
汤丽霞:电子科技大学生命科学与技术学院教授;研究领域:蛋白质工程、分子识别及核酸适配体相关研究。
林 昊:电子科技大学生命科学与技术学院教授;研究领域:生物信息学及人工智能在核酸适配体筛选与设计中的应用。
(作者:刘上华、刘入铭、汤丽霞、林昊)
(本文来源于公众号:生物化学与生物物理进展)
附件下载:
上一篇:
下一篇: